Publications
Citations
Journal Papers
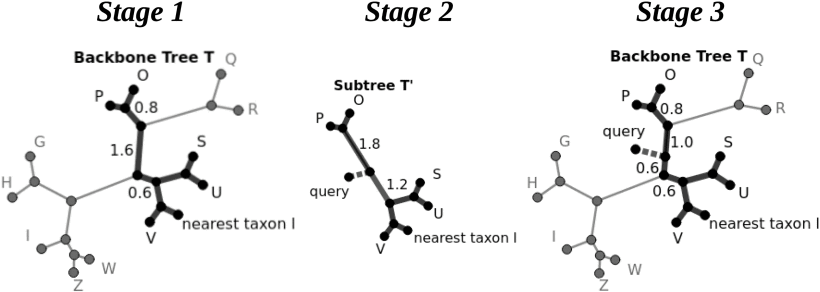
SCAMPP: Scaling Alignment-Based Phylogenetic Placement to Large Trees
Eleanor Wedell, Yirong Cai, Tandy Warnow
IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), 2023
Eleanor Wedell, Yirong Cai, Tandy Warnow
IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), 2023
Phylogenetic placement, the problem of placing a "query" sequence into a precomputed phylogenetic "backbone" tree, is useful for constructing large trees, performing taxon
identification of newly obtained sequences, and other applications. The most accurate current methods, such as pplacer and EPA-ng, are based on maximum likelihood and require
that the query sequence be provided within a multiple sequence alignment that includes the leaf sequences in the backbone tree. This approach enables high accuracy but also
makes these likelihood-based methods computationally intensive on large backbone trees, and can even lead to them failing when the backbone trees are very large (e.g., having
50,000 or more leaves). We present SCAMPP (SCaling AlignMent-based Phylogenetic Placement), a technique to extend the scalability of these likelihood-based placement methods
to ultra-large backbone trees. We show that pplacer-SCAMPP and EPA-ng-SCAMPP both scale well to ultra-large backbone trees (even up to 200,000 leaves), with accuracy that
improves on APPLES and APPLES-2, two recently developed fast phylogenetic placement methods that scale to ultra-large datasets. EPA-ng-SCAMPP and pplacer-SCAMPP are available
at https://github.com/cjehry04/PLUSplacer.
@article{wedell2022scampp,
title={SCAMPP: scaling alignment-based phylogenetic placement to large trees},
author={Wedell, Eleanor and Cai, Yirong and Warnow, Tandy},
journal={IEEE/ACM Transactions on Computational Biology and Bioinformatics},
volume={20},
number={2},
pages={1417--1430},
year={2022},
publisher={IEEE}
}
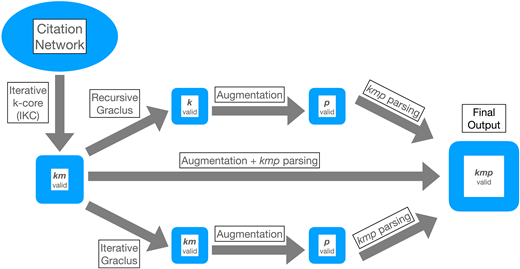
Center-periphery structure in research communities
Eleanor Wedell, Minhyuk Park, Dmitriy Korobskiy, Tandy Warnow, George Chacko
Quantitative Science Studies, 2022
Eleanor Wedell, Minhyuk Park, Dmitriy Korobskiy, Tandy Warnow, George Chacko
Quantitative Science Studies, 2022
Clustering and community detection in networks are of broad interest and have been the subject of extensive research that spans several fields. We are interested in
the relatively narrow question of detecting communities of scientific publications that are linked by citations. These publication communities can be used to identify
scientists with shared interests who form communities of researchers. Building on the well-known k-core algorithm, we have developed a modular pipeline to find
publication communities with center-periphery structure. Using a quantitative and qualitative approach, we evaluate community finding results on a citation network
consisting of over 14 million publications relevant to the field of extracellular vesicles. We compare our approach to communities discovered by the widely used
Leiden algorithm for community finding.
@article{wedell2022center,
title={Center--periphery structure in research communities},
author={Wedell, Eleanor and Park, Minhyuk and Korobskiy, Dmitriy and Warnow, Tandy and Chacko, George},
journal={Quantitative Science Studies},
volume={3},
number={1},
pages={289--314},
year={2022},
publisher={MIT Press One Broadway, 12th Floor, Cambridge, Massachusetts 02142, USA~…}
}
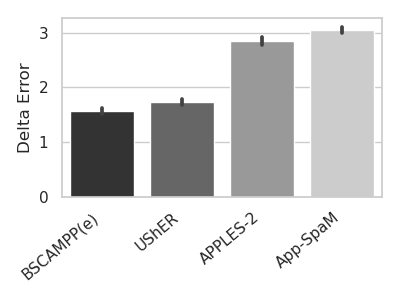
BSCAMPP: Batch-Scaled Phylogenetic Placement on Large Trees
Eleanor Wedell, Chengze Shen, Tandy Warnow
IEEE Transactions on Computational Biology and Bioinformatics, 2025
Eleanor Wedell, Chengze Shen, Tandy Warnow
IEEE Transactions on Computational Biology and Bioinformatics, 2025
Phylogenetic placement is the problem of placing sequences into a given phylogenetic tree, called a “backbone tree”. EPA-ng and pplacer are the two most accurate phylogenetic placement methods, but both can fail to complete when the backbone tree is very large. Our recently designed SCAMPP framework has been shown to scale both pplacer and EPA-ng to larger backbone trees of up to 180,000 sequences by building a small placement subtree for each query sequence and then using the phylogenetic placement method to place that query sequence into that subtree. However, the technique in SCAMPP produces many placement subtrees (potentially a different one for each query sequence), making it computationally expensive when placing many query sequences. Here we present BSCAMPP (Batch-SCAMPP), a new technique that overcomes this barrier by using the query sequences to select a much smaller number of placement subtrees. We show that BSCAMPP used with EPA-ng is much faster than SCAMPP used with EPA-ng, and scales to ultra-large backbone trees. We also show that BSCAMPP used with pplacer is much faster than SCAMPP used with pplacer, and somewhat more accurate but slower than BSCAMPP used with EPA-ng. BATCH-SCAMPP is freely available at https://github.com/ewedell/BSCAMPP.
@ARTICLE{10969524,
author={Wedell, Eleanor and Shen, Chengze and Warnow, Tandy},
journal={IEEE Transactions on Computational Biology and Bioinformatics},
title={BSCAMPP: Batch-Scaled Phylogenetic Placement on Large Trees},
year={2025},
volume={22},
number={4},
pages={1593-1605},
keywords={Phylogeny;Vegetation;Accuracy;Scalability;Runtime;Testing;Sequential analysis;Memory management;Maximum likelihood estimation;Training;Phylogenetic placement;EPA-ng;microbiome analysis;taxonomic identification;abundance profiling;pplacer},
doi={10.1109/TCBBIO.2025.3562281}}
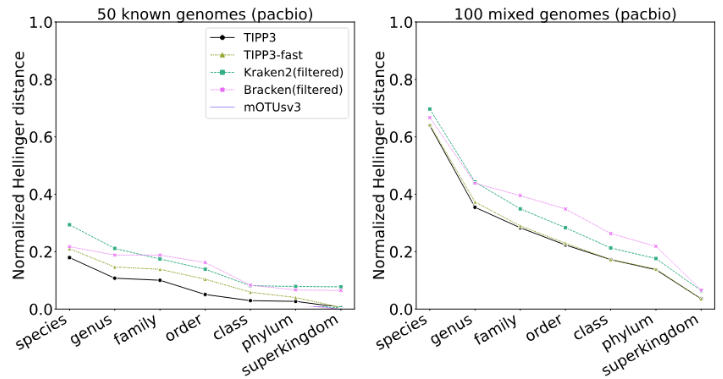
TIPP3 and TIPP3-fast: Improved Abundance Profiling in Metagenomics
Chengze Shen, Eleanor Wedell, Mihai Pop, Tandy Warnow
PLOS Computational Biology, 2025
Chengze Shen, Eleanor Wedell, Mihai Pop, Tandy Warnow
PLOS Computational Biology, 2025
We present TIPP3 and TIPP3-fast, new tools for abundance profiling in metagenomic datasets. Like its predecessor, TIPP2, the TIPP3 pipeline uses a maximum likelihood approach to place reads into labeled taxonomies using marker genes, but it achieves superior accuracy to TIPP2 by enabling the use of much larger taxonomies through improved algorithmic techniques. We show that TIPP3 is generally more accurate than leading methods for abundance profiling in two important contexts: when reads come from genomes not already in a public database (i.e., novel genomes) and when reads contain sequencing errors. We also show that TIPP3-fast has slightly lower accuracy than TIPP3, but is also generally more accurate than other leading methods and uses a small fraction of TIPP3’s runtime. Additionally, we highlight the potential benefits of restricting abundance profiling methods to those reads that map to marker genes (i.e., using a filtered marker-gene based analysis), which we show typically improves accuracy. TIPP3 is freely available at https://github.com/c5shen/TIPP3.
@article{10.1371/journal.pcbi.1012593,
doi = {10.1371/journal.pcbi.1012593},
author = {Shen, Chengze AND Wedell, Eleanor AND Pop, Mihai AND Warnow, Tandy},
journal = ,
publisher = {Public Library of Science},
title = ,
year = {2025},
month = {04},
volume = {21},
url = {https://doi.org/10.1371/journal.pcbi.1012593},
pages = {1-21},
number = {4},
}
Peer-Reviewed Conference Papers
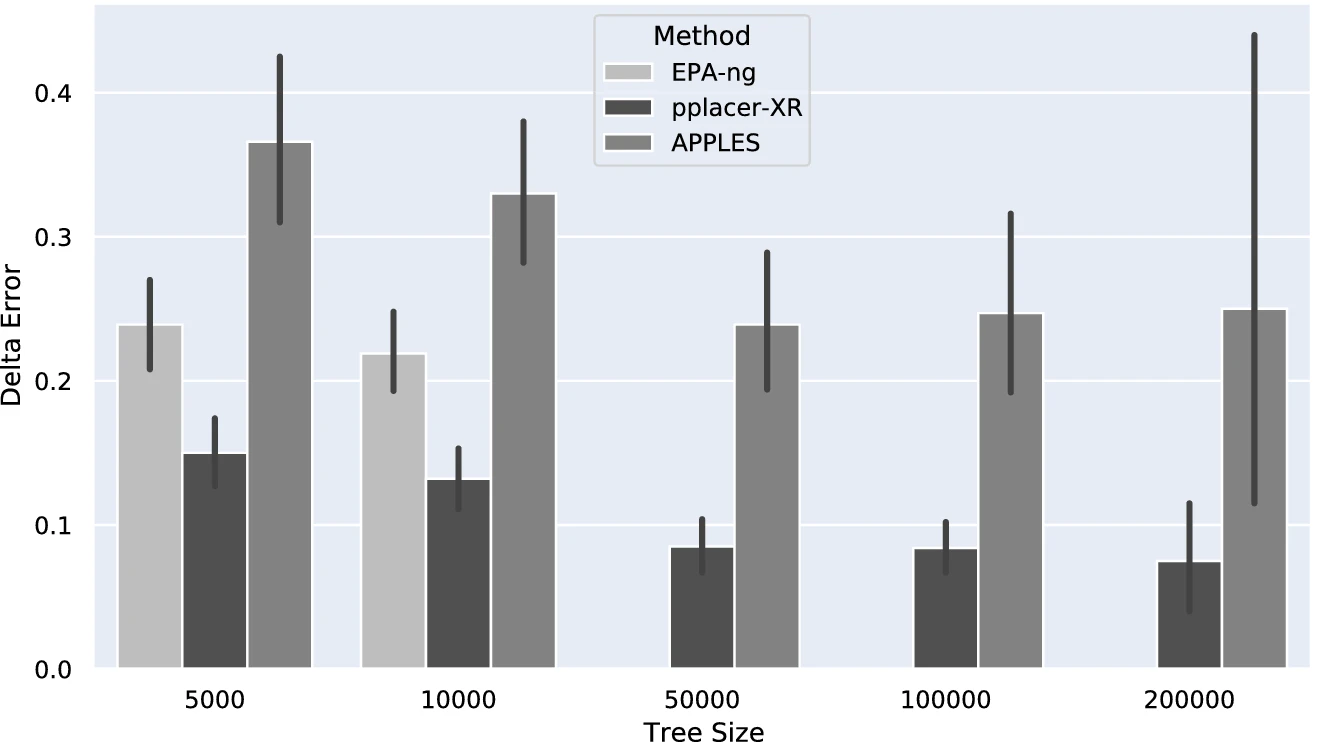
Scalable and Accurate Phylogenetic Placement Using pplacer-XR
Eleanor Wedell, Yirong Cai, Tandy Warnow
Algorithms for Computational Biology (AlCoB), 2021
Eleanor Wedell, Yirong Cai, Tandy Warnow
Algorithms for Computational Biology (AlCoB), 2021
Phylogenetic placement, the problem of placing a sequence into a precomputed phylogenetic “backbone” tree, is
useful for constructing large trees, performing taxon identification of newly obtained sequences, and other
applications. The most accurate current method, pplacer, performs the placement using maximum likelihood but
fails frequently on backbone trees with 5000 sequences. We show a simple technique, pplacer-XR (pplacer-eXtra
Range), that extends pplacer to large datasets. We show, using challenging large datasets, that pplacer-XR
provides the accuracy of pplacer and the scalability to ultra-large datasets of a leading fast phylogenetic
placmement method, APPLES. pplacer-XR is available in open source form on github.
@inproceedings{wedell2021scalable,
title={Scalable and accurate phylogenetic placement using pplacer-XR},
author={Wedell, Eleanor and Cai, Yirong and Warnow, Tandy},
booktitle={International Conference on Algorithms for Computational Biology},
pages={94--105},
year={2021},
organization={Springer}
}
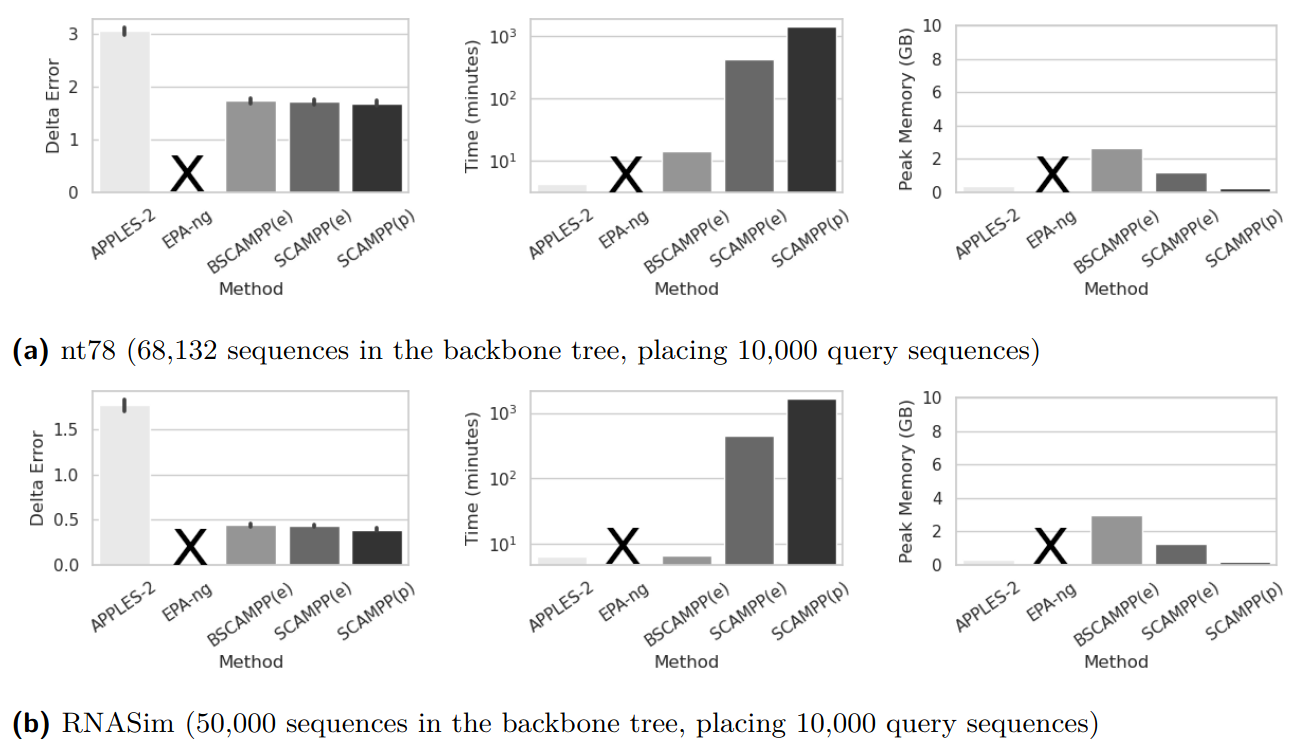
BATCH-SCAMPP: Scaling phylogenetic placement methods to place many sequences (Abstract)
Eleanor Wedell, Chengze Shen, Tandy Warnow
In 23rd International Workshop on Algorithms in Bioinformatics (WABI), 2023
Eleanor Wedell, Chengze Shen, Tandy Warnow
In 23rd International Workshop on Algorithms in Bioinformatics (WABI), 2023
Phylogenetic placement is the problem of placing one or more query sequences into a phylogenetic "backbone"
tree, which may be a maximum likelihood tree on a multiple sequence alignment for a single gene, a taxonomy
with leaves labeled by sequences for a single gene [Nidhi Shah et al., 2021], or a species tree [Jiang et al.,
2023]. When the backbone tree is a tree estimated on a single gene, the most accurate techniques for
phylogenetic placement are likelihood-based, and can be computationally intensive when the backbone trees are
large [Chu and Warnow, 2023]. Phylogenetic placement into gene trees occurs when updating existing gene trees
with newly observed sequences, but can also be applied in the "bulk" context, where many sequences are placed
at the same time into the backbone tree. For example, phylogenetic placement can be used to taxonomically
characterize shotgun sequencing reads generated for an environmental sample in metagenomic analysis [Nidhi
Shah et al., 2021; Barbera et al., 2019]. The two most well known maximum likelihood phylogenetic placement
methods are pplacer [Chu and Warnow, 2023] and EPA-ng [Barbera et al., 2019]. Of these two, EPA-ng is
optimized for scaling the number of query sequences and is capable of placing millions of sequences into
phylogenetic trees of up to a few thousand sequences [Barbera et al., 2019], and achieves sublinear runtime
in the number of query sequences (see Figure 2 from [Balaban et al., 2022]). Previously we introduced the
SCAMPP framework [Wedell et al., 2022] to enable both pplacer and EPA-ng to perform phylogenetic placement
into ultra-large backbone trees, and we demonstrated its utility for placing into backbone trees with up to
200,000 sequences. By using maximum likelihood methods pplacer or EPA-ng within the SCAMPP framework, the
resulting placements are more accurate than with APPLES-2 [Balaban et al., 2022], with the most notable
accuracy improvement for fragmentary sequences, and are computationally similar for single query sequence
placement [Wedell et al., 2022]. However, SCAMPP was designed to incrementally update a large tree, one
query sequence at a time, and was not optimized for the other uses of phylogenetic placement, where batch
placement of many sequencing reads is required. Here we introduce BATCH-SCAMPP, a technique that improves
scalability in both dimensions: the number of query sequences being placed into the backbone tree and the
size of the backbone tree. Furthermore, BATCH-SCAMPP is specifically designed to improve EPA-ng's
scalability to large backbone trees. Although BATCH-SCAMPP is based on SCAMPP, it uses a substantially
modified design in order to be able to take advantage of EPA-ng's ability to place many query sequences
efficiently. The BATCH-SCAMPP method operates by allowing the input set of query sequences to suggest and
then vote on placement subtrees, thus enabling many query sequences to select the same placement subtree.
We pair BATCH-SCAMPP with EPA-ng to explore the capability of this approach for scaling to many query
sequences. We show that this combination of techniques (which we call BSCAMPP+EPA-ng, or BSCAMPP(e)) not
only provides high accuracy and scalability to large backbone trees, matching that of SCAMPP used with
EPA-ng (i.e., SCAMPP(e)), but also achieves the goal of scaling sublinearly in the number of query
sequences. Moreover, it is much more scalable than EPA-ng and faster than SCAMPP+EPA-ng: when placing
10,000 sequences into a backbone tree of 50,000 leaves, EPA-ng is unable to run due to memory issues,
SCAMPP+EPA-ng requires 1421 minutes, and BSCAMPP(e) places all sequences in 7 minutes (all given the
same computational resources). Figure 1 gives an example of this performance advantage on the nt78 [Chu
and Warnow, 2023] simulated dataset.
@InProceedings{wedell_et_al:LIPIcs.WABI.2023.3,
author = {Wedell, Eleanor and Shen, Chengze and Warnow, Tandy},
title = ,
booktitle = {23rd International Workshop on Algorithms in Bioinformatics (WABI 2023)},
pages = {3:1--3:2},
series = {Leibniz International Proceedings in Informatics (LIPIcs)},
ISBN = {978-3-95977-294-5},
ISSN = {1868-8969},
year = {2023},
volume = {273},
editor = {Belazzougui, Djamal and Ouangraoua, A\"{i}da},
publisher = {Schloss Dagstuhl -- Leibniz-Zentrum f{\"u}r Informatik},
address = {Dagstuhl, Germany},
URL = {https://drops.dagstuhl.de/entities/document/10.4230/LIPIcs.WABI.2023.3},
URN = {urn:nbn:de:0030-drops-186296},
doi = {10.4230/LIPIcs.WABI.2023.3},
annote = {Keywords: Phylogenetic Placement, EPA-ng, Phylogenetics}
}
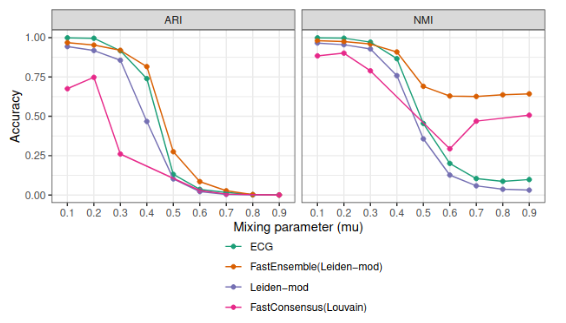
FastEnsemble: A New Scalable Ensemble Clustering Method
Yasamin Tabatabaee, Eleanor Wedell, Minhyuk Park, Tandy Warnow
Complex Networks and their Applications (CNA), 2024
Yasamin Tabatabaee, Eleanor Wedell, Minhyuk Park, Tandy Warnow
Complex Networks and their Applications (CNA), 2024
Many community detection algorithms are stochastic in nature, and their output can vary based on different input
parameters and random seeds. Consensus clustering methods, such as FastConsensus and ECG, combine clusterings
from multiple runs of the same clustering algorithm, in order to improve stability and accuracy. In this study
we present a new consensus clustering method, FastEnsemble, and show that it provides advantages over both
FastConsensus and ECG. Furthermore, FastEnsemble is designed for use with any clustering method, and we show
results using \ourmethod with Leiden optimizing modularity or the Constant Potts model. FastEnsemble is
available in Github at https://github.com/ytabatabaee/fast-ensemble.
@InProceedings{10.1007/978-3-031-82435-7_5,
author="Tabatabaee, Yasamin
and Wedell, Eleanor
and Park, Minhyuk
and Warnow, Tandy",
editor="Cherifi, Hocine
and Donduran, Murat
and Rocha, Luis M.
and Cherifi, Chantal
and Varol, Onur",
title="FastEnsemble: A New Scalable Ensemble Clustering Method",
booktitle="Complex Networks {\&} Their Applications XIII",
year="2025",
publisher="Springer Nature Switzerland",
address="Cham",
pages="57--70",
isbn="978-3-031-82435-7"
}
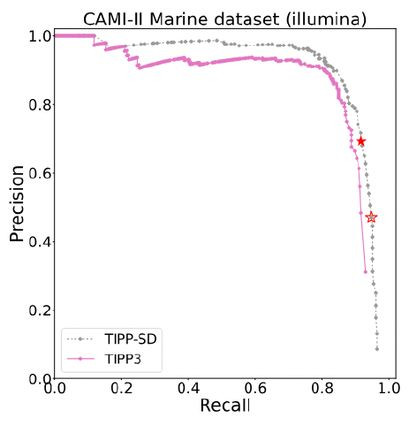
In this study, we present TIPP-SD (i.e., TIPP for Species Detection), a new technique for species detection in a microbiome sample. TIPP-SD uses a modified version of TIPP3, which is a recently developed abundance profiling tool based on maximum likelihood phylogenetic placement into marker gene taxonomies. TIPP-SD depends on a parameter (i.e., threshold) for the required support for species detection, thus allowing us to compute a precision-recall curve as we vary this parameter. In comparing the precision-recall curves for TIPP-SD, TIPP3, Kraken2, Bracken, and Metapresence, we find that TIPP-SD improves on the other methods with respect to accuracy under conditions where some species occur in low abundance, or where there is sequencing error. Under easier conditions, TIPP-SD is close to the best of these methods. Finally, although TIPP-SD is slower than Kraken2 and Bracken, it is still fast enough to be used on large datasets.
@article {Shen2025.08.27.672749,
author = {Shen, Chengze and Wedell, Eleanor and Pop, Mihai and Warnow, Tandy},
title = {TIPP-SD: A New Method for Species Detection in Microbiomes},
elocation-id = {2025.08.27.672749},
year = {2025},
doi = {10.1101/2025.08.27.672749},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2025/09/01/2025.08.27.672749},
eprint = {https://www.biorxiv.org/content/early/2025/09/01/2025.08.27.672749.full.pdf},
journal = {bioRxiv}
}
Masters Thesis
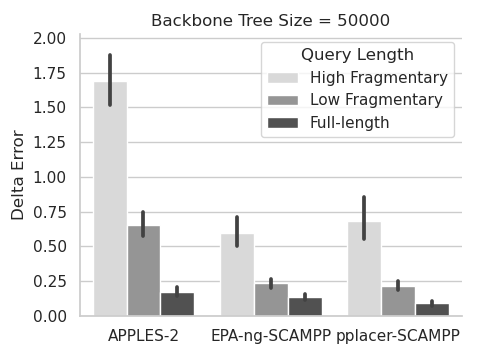
SCAMPP: Scalable alignment-based phylogenetic placement
Eleanor Wedell Advisor: Professor Tandy Warnow
Master thesis. University of Illinois Urbana-Champaign, 2022
Eleanor Wedell Advisor: Professor Tandy Warnow
Master thesis. University of Illinois Urbana-Champaign, 2022
Phylogenetic placement, the problem of placing a "query" sequence into a precomputed phylogenetic "backbone” tree, is useful for constructing large trees,
performing taxon identification of newly obtained sequences, and other applications. The most accurate current methods, such as pplacer and EPA-ng, are
based on maximum likelihood and require that the query sequence be provided within a multiple sequence alignment that includes the leaf sequences in the
backbone tree. This approach enables high accuracy but also makes these likelihood-based methods computationally intensive on large backbone trees, and
can even lead to them failing when the backbone trees are very large (eg, having 50,000 or more leaves). We present SCAMPP (SCAlable alignMent-based
Phylogenetic Placement), a technique to extend the scalability of these likelihood-based placement methods to ultra-large backbone trees. We show that
pplacer-SCAMPP and EPA-ng-SCAMPP both scale well to ultra-large backbone trees (even up to 200,000 leaves), with accuracy that improves on APPLES and
APPLES-2, two recently developed fast phylogenetic placement methods that scale to ultra-large datasets. EPA-ng-SCAMPP and pplacer-SCAMPP are available
at https://github.com/chry04/PLUSplacer.
@phdthesis{wedell2022scampp,
title={SCAMPP: Scalable alignment-based phylogenetic placement},
author={Wedell, Eleanor},
year={2022},
school={University of Illinois at Urbana-Champaign}
}